



遗传性痉挛性截瘫是一种比较少见的家族遗传性疾病,最常见为常染色体显性遗传,也有常染色体隐性遗传及X连锁隐性遗传。以慢性进行性无力与慢性痉挛性下肢瘫痪为特征。解开其发病机制一直是生命科学家的心愿。
日前,《Movement Disorders》期刊在线发表了题为《A Novel SPAST Mutation Results in Spastin Accumulation and Defects in Microtubule Dynamics》的研究论文,该研究由中国科学院脑科学与智能技术卓越创新中心(神经科学研究所)、神经科学国家重点实验室刘静宇研究组完成。

该研究通过对遗传病家系的连锁分析和Sanger测序,并结合细胞系模型分析,发现遗传性痉挛性截瘫(HSP)致病基因SPAST编码的截短蛋白质spastin可以通过异构体特异性的方式干扰微管的动态平衡,进而导致HSP的发生。此研究进一步提出spastin的截短突变体可能通过长期的细胞积累方式影响皮质脊髓束的功能,为探究遗传性痉挛性截瘫4型(SPG4)的致病机制和开展治疗提供了新的方向。
遗传性痉挛性截瘫是一种遗传和临床异质性神经退行性疾病,其主要特征是下肢进行性痉挛和无力。由SPAST基因突变引起的痉挛性截瘫4型是最常见的常染色体显性遗传的HSP亚型。目前普遍认为该基因突变导致的蛋白质功能丧失引起单倍量不足(haploinsufficiency)是该疾病的致病机制,但该学说并不能很好地阐明痉挛性截瘫的临床表型和突变蛋白质功能损伤程度之间的相关性,并且相应的临床干预治疗进展也十分缓慢。
SPAST基因主要编码M1(68 kDa)和M87(60 kDa)两种异构体并发挥微管剪切活性,维持微管的动态平衡。刘静宇研究组前期收集了六个自然村的三个大的遗传性痉挛性截瘫家系(247名成员,共67名患者)(图1),通过连锁分析将三个家系的致病区段同时定位到SPAST基因座,进一步的Sanger测序证实所有病人均携带SPAST的一个新的插入突变c.985dupA(p.Met329Asnfs*3),该突变产生两种截短异构体dupA-M1和dupA-M87,这两种突变体的蛋白质降解速率降低。
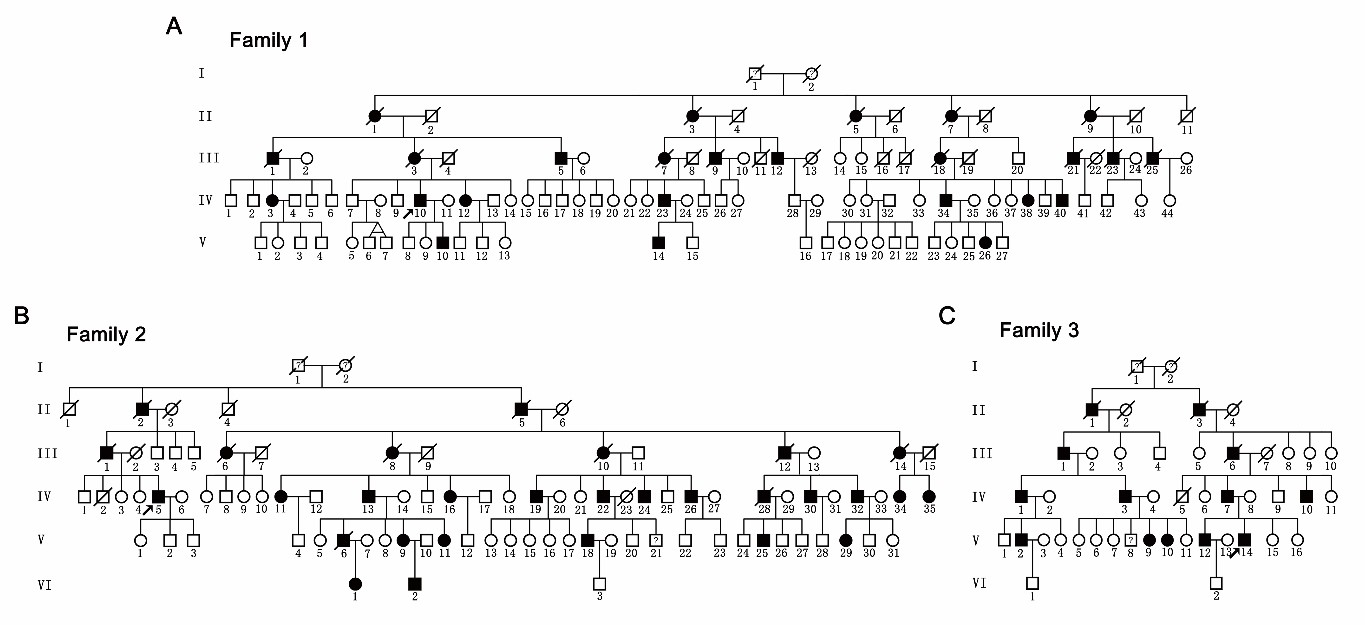
图1 三个来自同一个祖先的常染色体显性遗传的痉挛性截瘫家系系谱
与此同时,还发现dupA-M1与微管紧密结合(细胞内纤维状分布),并且阻断了微管的解聚过程;而dupA-M87却均匀地分布于胞质与细胞核中,并未显示出对微管解聚的干扰(图2A)。
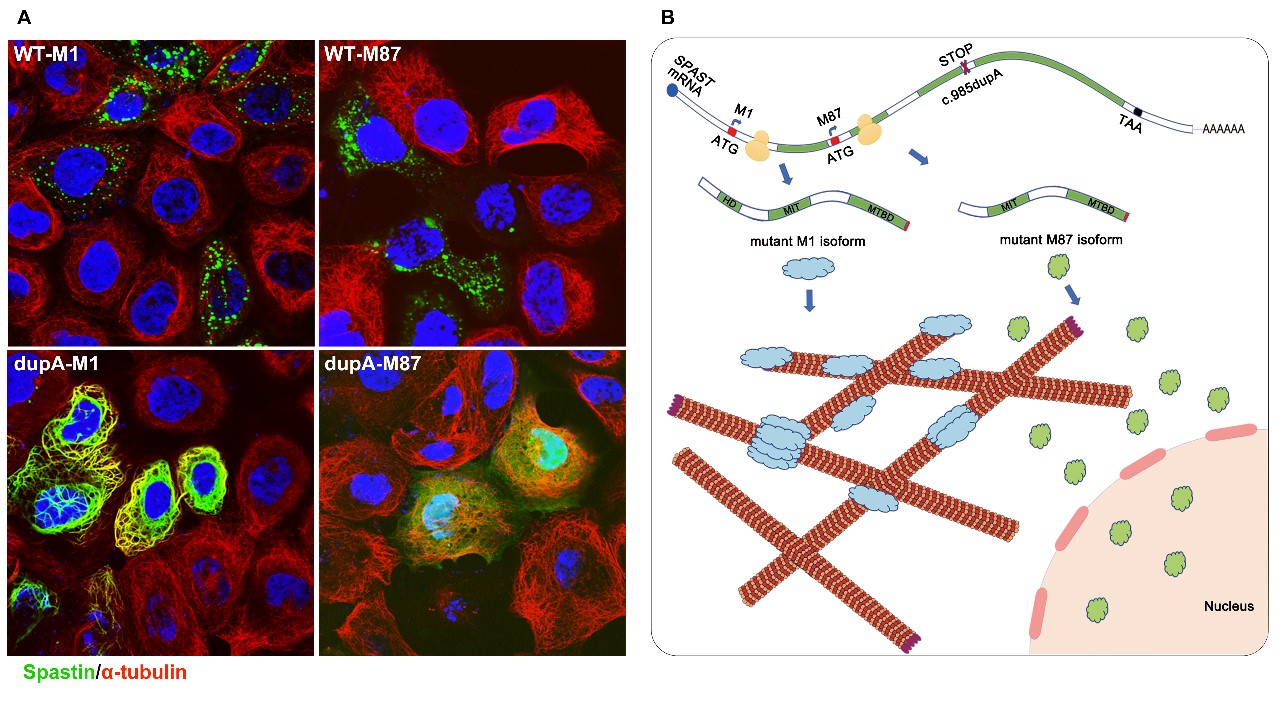
图2(A)野生型(WT-M1和WT-M87)和截短体(dupA-M1和dupA-M87)蛋白质的细胞定位。(B)spastin突变体c.985dupA(p.Met329Asnfs*3)的致病机制假说。
该研究发现SPAST突变导致的spastin截短体可以在细胞内长时程积累,因此可能导致高表达spastin的皮质脊髓束及其远端轴突中产生细胞毒性。
此外,该研究一定程度上揭示了突变体spastin可能通过异构体特异性的方式影响皮质脊髓束的功能,从而导致遗传性痉挛性截瘫疾病(图2B)。但是其具体的作用机制还有待进一步探索。
华中科技大学生命科学与技术学院博士研究生陈瑞、杜士月和湖北省妇幼保健院医学遗传学中心姚妍怡为共同第一作者,在刘静宇研究员和博士后徐旋的指导下完成,刘静宇研究组的其他成员积极参与,是众多师生合作的重要成果。
作者:许琦敏
图源:受访者提供
责任编辑:任荃
*文汇独家稿件,转载请注明出处。